
¿QUÉ ES DYRK1A?
DYRK1A es el nombre de un gen localizado en el brazo largo del cromosoma 21, en la región q22.13. Este gen codifica la enzima quinasa 1A regulada por fosforilación de tirosina de especificidad dual, que juega un importante papel en el desarrollo del sistema nervioso.
¿QUÉ ES EL SÍNDROME DYRK1A?
El síndrome DYRK1A es una enfermedad caracterizada por una serie de signos y síntomas que incluyen: discapacidad intelectual, problemas en el lenguaje, dificultades de alimentación, epilepsia, alteración conductual dentro del trastorno del espectro autista, tamaño reducido de la cabeza (microcefalia) y rasgos faciales particulares entre otros.
¿QUÉ CAUSA EL SÍNDROME DYRK1A?
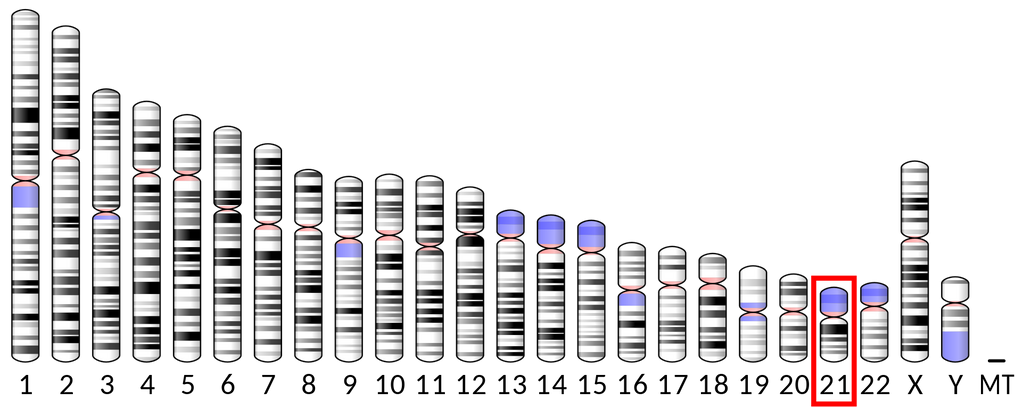
El síndrome DYRK1A se debe a cambios genéticos que ocurren en el gen DYRK1A y que afectan a la función de la proteína codificada por el mismo. Estos cambios genéticos se denominan variantes o mutaciones. Este gen tiene un papel importante en la regulación de la función de muchas otras proteínas en la célula.
¿CÓMO SE DIAGNOSTICA EL SÍNDROME DYRK1A?
Los cambios en DYRK1A se pueden identificar mediante estudios genéticos moleculares. El cariotipo molecular o arrayCGH permite detectar deleciones o duplicaciones que impliquen al gen DYRK1A. Cuando se trata de variantes puntuales, el gen DYRK1A se encuentra incluido en paneles NGS (next generation sequencing) para el estudio de pacientes con cuadros de discapacidad intelectual, microcefalia, autismo y epilepsia. También la secuenciación completa del exoma o del genoma permite detectar cambios en el gen DYRK1A. Es fundamental que estos estudios se precedan de un correcto asesoramiento pretest y que los resultados sean interpretados y explicados a las familias por un genetista.
¿DIFERENTES VARIANTES CAUSAN DIFERENTES SÍNTOMAS Y GRAVEDAD?
No hay evidencia hasta la fecha de que exista correlación entre el tipo de variante ni su localización en el gen, con la gravedad de las manifestaciones clínicas en los afectados. Probablemente, otros factores genéticos y ambientales influyan en la expresividad clínica variable para esta enfermedad.
¿ES LA DELECCIÓN 21q22.13 LO MISMO?
Sí, DYRK1A es el gen responsable de las manifestaciones observadas en los pacientes portadores de una deleción (pérdida de material genético) de esta región 21q22.13. Los pacientes con deleción 21q22.13 y aquellos con variantes puntuales en el gen DYRK1A presentan manifestaciones clínicas similares.
¿QUÉ RELACIÓN HAY ENTRE SÍNDROME DYRK1A Y SÍNDROME DE DOWN?
Las personas suelen nacer con 23 pares de cromosomas, 46 cromosomas en total. En el síndrome de Down, hay tres copias del cromosoma 21 y, por tanto, tres copias del gen DYRK1A. Al haber 3 copias del gen DYRK1A existe un exceso de proteína DYRK1A. Por este motivo, un objetivo terapéutico en el síndrome de Down sería la inhibición o disminución de este exceso de proteína. Por el contrario, en el síndrome de haploinsuficiencia de DYRK1A, aunque hay un par de cromosomas 21, el gen DYRK1A de uno de ellos es deficitario o defectuoso.
¿QUÉ SE DEBE ESPERAR DEL DESARROLLO DE UN AFECTADO?
Cada persona con este diagnóstico es única y puede no desarrollar la totalidad de las características descritas para la enfermedad. Es frecuente el retraso en el desarrollo psicomotor, las dificultades importantes en la comunicación y el lenguaje y la epilepsia de inicio en la primera infancia, que puede o no mantenerse en la adolescencia y hasta la edad adulta. También son comunes los problemas de conducta como la ansiedad o las estereotipias. Pueden existir problemas de alimentación desde el nacimiento, con dificultad para coordinar la succión con la deglución y la respiración, así como aversión a las texturas.
¿HAY ACTUALMENTE UNA CURA?
Actualmente no existe una cura para el síndrome DYRK1A.
¿CUALES SON LOS TRATAMIENTOS?
La Atención Temprana, la logopedia, terapia ocupacional y física, pueden ayudar a los afectados a alcanzar su máximo potencial. Los pacientes precisan de una atención interdisciplinar individualizada y de por vida.
¿CUÁL ES LA PREVALENCIA DE DYRK1A?
Es difícil saber el número exacto de casos porque no existe un registro formal. Sin embargo, se estima que alrededor del 0,3 % al 0,5 % de las personas con discapacidad intelectual se encuentran en realidad afectadas por esta enfermedad. El número de pacientes diagnosticados aumentará sin duda en los próximos años a medida que exista un mayor acceso a pruebas genéticas.
SI DESEAS CONOCER MÁS ACERCA DE DYRK1A, NO DUDES EN CONTACTAR CON NOSOTROS O INSCRÍBETE Y COLABORA JUNTO CON LAS FAMILIAS AFECTADAS EN EL AVANCE DE LA INVESTIGACIÓN Y DIVULGACIÓN DE ESTE SÍNDROME ULTRARRARO, TE ESPERAMOS.
MÁS DE 630 CASOS DIAGNOSTICADOS EN 51 PAÍSES DE TODO EL MUNDO
CON ALREDEDOR DE 21 CASOS EN ESPAÑA
Copyright © Todos los derechos reservados · Inscrita en el Registro Nacional de Asociaciones Sección 1ª Número 624373 · CIF: G-09701962








